Dijun Chen’s team have developed the SeiPlant, a deep-learning framework for cross-species prediction of plant histone modifications
Nanjing, China — A research team from Nanjing University, in collaboration with Zhejiang University, has introduced SeiPlant, a deep-learning framework that predicts plant histone modification landscapes directly from DNA sequence and systematically tests how well such models transfer across species.
Why this matters
Histone modifications (such as H3K4me3 and H3K27ac) are core components of plant gene regulation, shaping chromatin accessibility and transcriptional activity. Yet high-quality epigenomic profiles (e.g., ChIP-seq) are still sparse for most non-model and many agriculturally important plants. In their study, the team asks a practical question for plant functional genomics: can a model trained in one species accurately predict histone modifications in another—and what training strategy best supports generalization?
A four-stage, plant-focused framework for cross-species epigenomic prediction
SeiPlant adapts the Sei deep-learning architecture into a plant-oriented, multi-stage benchmarking framework that spans (i) species-specific modeling, (ii) cross-species transfer, (iii) within-family generalization, and (iv) cross-family evaluation. Trained on plant chromatin profiling data for major histone marks (compiled from ChIP-Hub and curated into high-confidence regulatory intervals), SeiPlant uses ~1 kb DNA sequence windows as input to generate genome-wide chromatin feature predictions, providing a practical route to annotate epigenomic patterns directly from sequence across diverse plant species.
Strong within-species accuracy—and a clear “phylogeny effect” across species
When trained and evaluated within the same species, SeiPlant delivered strong predictive accuracy across three representative plant genomes—rice (Oryza sativa), Arabidopsis thaliana, and maize (Zea mays)—under rigorous held-out chromosome testing (rice AUROC 0.970 / AUPRC 0.754, Arabidopsis 0.941 / 0.714, maize 0.965 / 0.718). Beyond these summary metrics, the predicted genome-wide signal tracks closely matched experimental ChIP-seq profiles in representative genomic regions, supporting biological realism at locus-level resolution. In contrast, cross-species transfer revealed a clear limitation: performance declined with increasing evolutionary distance—most noticeably between monocots and dicots—underscoring the need for phylogeny-aware strategies rather than naïve model deployment across divergent lineages.
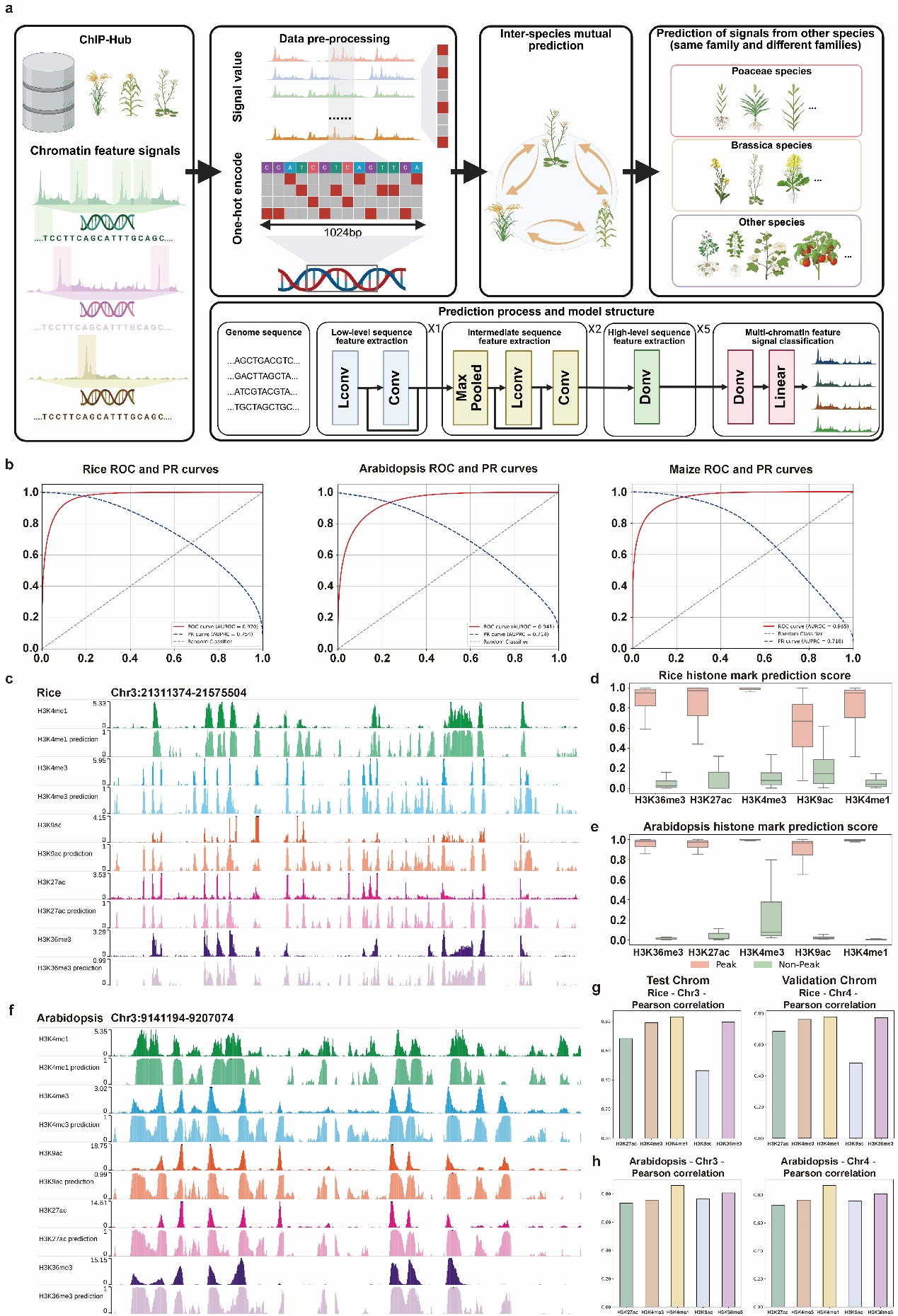
Figure 1. Framework and performance of species-specific chromatin feature prediction in plants. (a) Overview of the cross-species chromatin feature prediction framework. (b) Receiver operating characteristic (ROC) and precision-recall (PR) curves for species-specific models in Rice, Arabidopsis, and Maize. ROC curves are shown in red; PR curves are shown in blue. (c) Predicted and experimental signal tracks for representative histone marks in Rice (Chr3:21311374–21575504). (d) The distribution Boxplot of prediction scores for peak and non-peak regions across five histone marks in Rice. (e) The distribution Boxplot of prediction scores for peak and non-peak regions across five histone marks in Arabidopsis. (f) Predicted and experimental signal tracks for representative histone marks in Arabidopsis (Chr3:9141194–9207074). (g) Pearson correlation coefficients between predicted and experimental signals across test and validation chromosomes for each histone mark in Rice. (h) Pearson correlation coefficients between predicted and experimental signals across test and validation chromosomes for each histone mark in Arabidopsis.
Family-level training improves generalization to unprofiled crops
A key takeaway from the study is that phylogenetically informed training can materially improve transfer. The authors built a Poaceae family-level model by jointly training on rice + maize, and then evaluated it in other grasses (including Setaria italica, Sorghum bicolor, Brachypodium distachyon, and Hordeum vulgare).
In cross-species tests, models trained on closer relatives consistently outperformed those trained on more distant species. For example, for rice H3K4me3 prediction, a maize-trained model achieved higher correlations than an Arabidopsis-trained model in representative regions (maximum Pearson correlation 0.72 vs 0.52, respectively).
Overall, the work supports a practical strategy for the community: train within phylogenetic families when possible (e.g., Poaceae, Brassicaceae) to achieve more reliable genome-wide epigenomic annotation in species that lack experimental profiling.
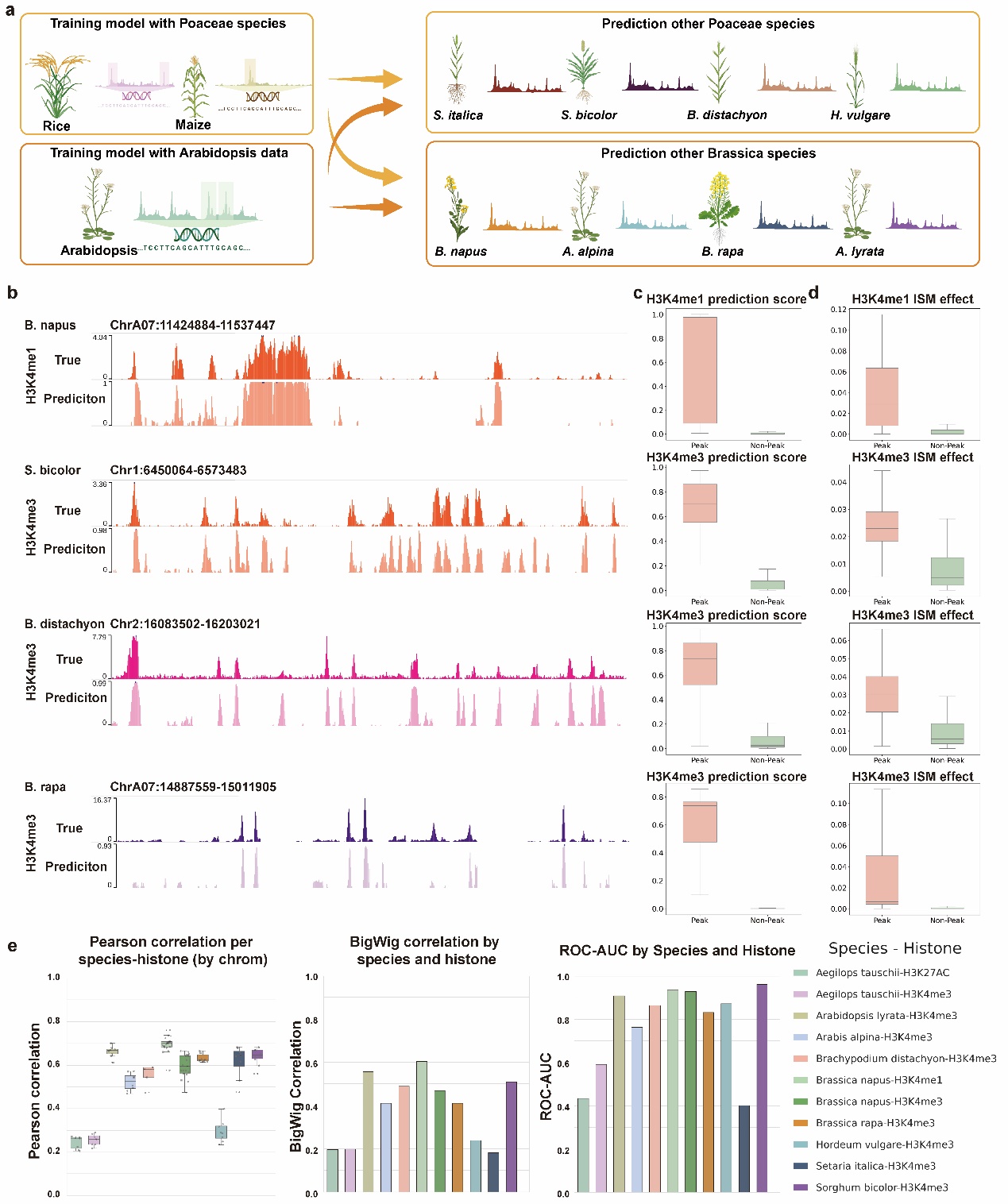
Figure 3. Family-level models improve cross-species prediction of chromatin features. (a) Schematic illustration of family-level training and prediction: the Poaceae model trained on Rice and Maize was used to predict signals in other Poaceae species; the Arabidopsis-trained model was used to predict signals in other Brassicaceae species. (b) Comparison of predicted and observed histone modification signals across representative genomic intervals in multiple Poaceae and Brassicaceae species. (c) Boxplots showing prediction scores for peak and non-peak regions across three histone modifications. (d) Boxplots showing ISM-based Δ scores for peak and non-peak regions across three histone modifications. (e) Quantitative evaluation of family-level prediction performance using Pearson correlation, BigWig correlation, and ROC-AUC for each species-histone pair.
An easy-to-use pipeline for genome-wide chromatin signal prediction
Beyond benchmarking, the authors provide an easy-to-use pipeline that enables researchers to generate genome-wide chromatin signal predictions directly from DNA sequence, helping lower the experimental and computational barriers to functional annotation—especially for non-model plant species where epigenomic profiling data remain limited.
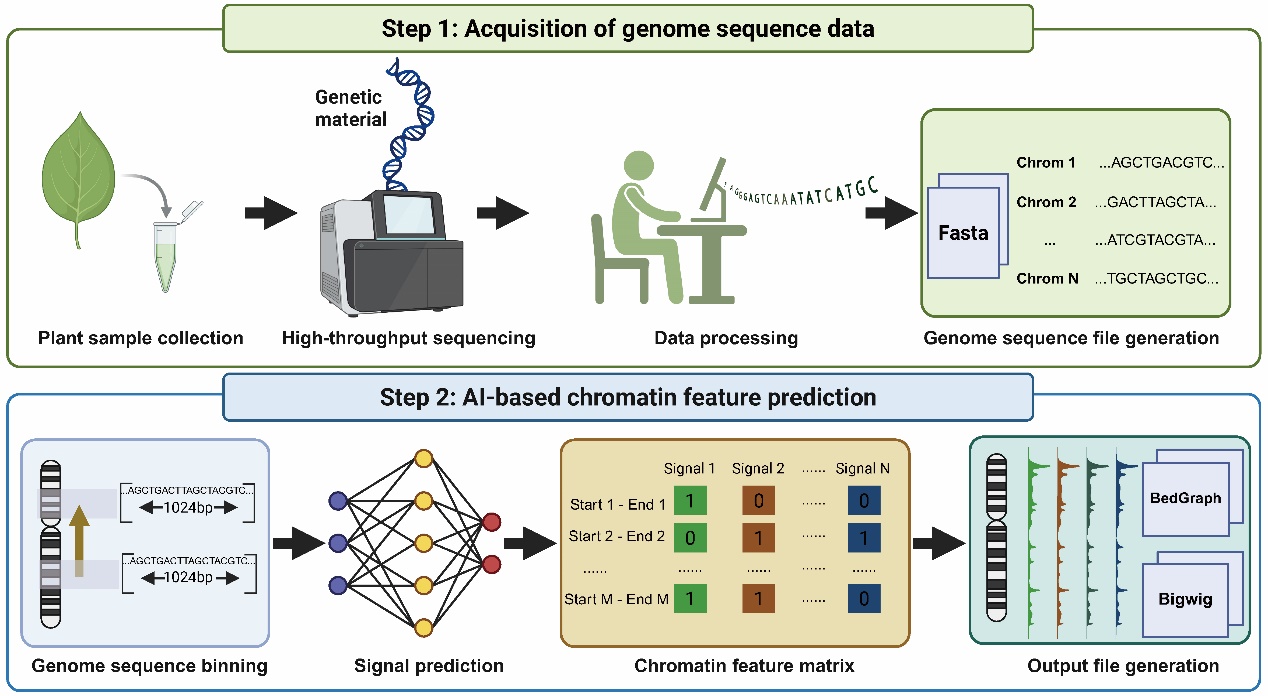
Figure 7. User-friendly pipeline for chromatin signal prediction from genomic sequences. In Step 1, plant samples are collected and sequenced using next-generation sequencing (NGS) to obtain genomic data, which is processed into a standardized FASTA file containing chromosome-level sequences. In Step 2, the genome is binned into fixed-length intervals, and the AI-based prediction model is applied to infer the presence and intensity of histone modification signals.
Reference: Lv, T., Han, Q., Li, Y. et al. Cross-species prediction of histone modifications in plants via deep learning. Genome Biol (2026). https://doi.org/10.1186/s13059-025-03929-4.