Cell Genomics | Dijun Chen's Team Develops SSpMosaic: A Universal Framework for Single-Cell and Spatial Omics Integration and Annotation via Metaprograms
Single-cell and spatial omics technologies are transforming our ability to map cellular composition and tissue architecture. However, integrating and interpreting large-scale datasets across batches, modalities, and species remains a major bottleneck, and many existing workflows rely on fragmented toolchains or “black-box” models with limited biological interpretability.
To address these challenges, a team led by Dijun Chen from the School of Life Sciences, Nanjing University, has developed SSpMosaic, a novel computational framework published in Cell Genomicson December 19, 2025. SSpMosaic introduces metaprograms (MPs) as cross-dataset conserved gene program anchors, providing a consistent biological “language” that supports downstream tasks in a one-stop workflow—from multi-omics integration and cell annotation to spatial deconvolution and cross-slice spatial characterization.
How SSpMosaic works. SSpMosaic begins by extracting gene co-expression or co-regulation modules from each dataset to form dataset-specific gene programs. It then uses network propagation to quantify similarity among programs across datasets and applies hierarchical clustering to merge them into cross-sample conserved metaprograms. These MPs capture recurrent and interpretable molecular functions and states, they serve as stable anchors for aligning heterogeneous datasets and for consistent interpretation across experiments, platforms, and species (Figure 1). Based on this foundation, SSpMosaic achieves:
1. Robust Multi-omics Integration: Seamlessly aligns data across batches, modalities, and species—exemplified by the successful integration of human-mouse brain atlases and challenging tri-modal data (transcriptome, epigenome, proteome) while preserving distinct biological signals.
2. High-Precision Annotation: Accurately annotates cell types and discovers novel cell states. It outperforms existing methods by precisely discriminating subtle neuronal subtypes and automatically identifying novel cell populations in large-scale cross-tissue atlases.
3. Resolution-Agnostic Deconvolution: Works from spot-level to subcellular resolutions. SSpMosaic restores spatial architectures and functional domains across mouse olfactory bulb (10x Visium, spot level), hippocampus (10x Visium HD, subcellular level), and human non-small cell lung cancer tissues (CosMx, single-cell level), consistently outperforming state-of-the-art methods.
4. Importantly—Reference-Free Spatial Analysis: Identifies recurrent spatial structures directly from spatial data, breaking the critical dependency on single-cell references.
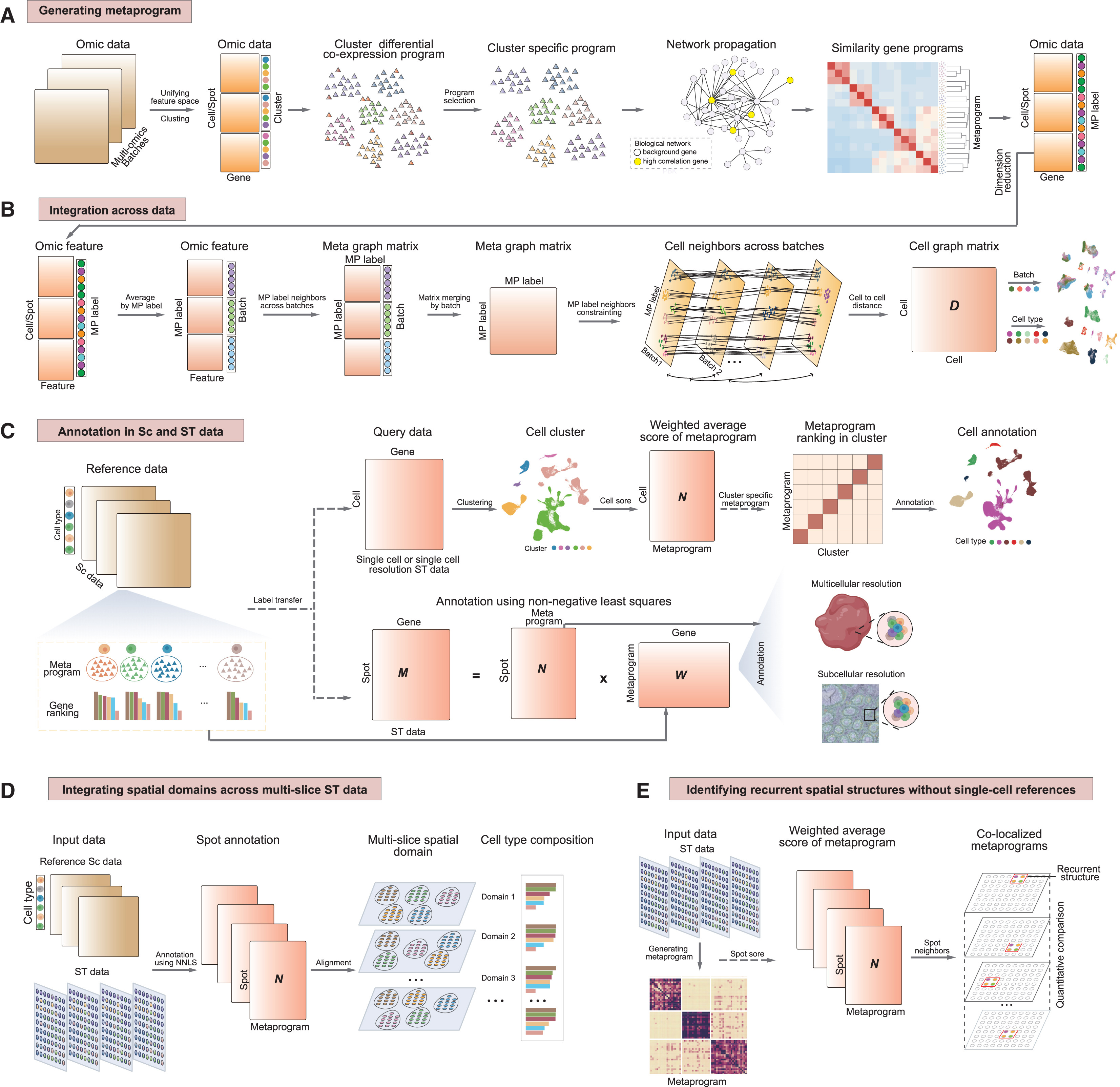
Figure 1. Schematic overview of SSpMosaic. (A) Construction of metaprograms across single-cell and spatial omics datasets. (B) Workflow of data integration through the anchors of metaprograms. (C) Cell/spatial annotation using reference metaprograms in (A). (D) Cross-slice spatial transcriptomics analysis. (E) Reference-free spatial characterization.
Highlight: reference-free discovery of recurrent GBM spatial structures.
A striking application of SSpMosaic is its reference-free spatial analysis of glioblastoma (GBM). In an analysis of 26 GBM spatial transcriptomics slices without paired single-cell data, SSpMosaic inferred 17 metaprograms directly from spatial measurements (Figure 2). The framework not only recovered known cellular programs but also highlighted additional states, including tumor-associated macrophage (TAM)–related signals.
To quantify a recurrent spatial structure in which TAM-associated regions are “wrapped” by hypoxic regions, the study introduced a novel metric, the Encapsulation Index. This enabled stratification of slices into groups with high versus low TAM–hypoxia encapsulation and supported downstream analyses of spatially organized immunosuppressive signaling pathways. Together, these results demonstrate that SSpMosaic can reveal reproducible, higher-order spatial organization and provide mechanistic insights into tumor microenvironment architecture—even in the absence of matched single-cell references.

Figure 2. Integration of multi-slice GBM spatial transcriptomics and identification of recurrent spatial structures without scRNA reference. (A) Schematic workflow for integrating multi-slice GBM spatial transcriptomics data and identifying recurrent spatial structures. (B) Heatmap showing similarity of metaprograms identified by the SSpMosaic algorithm across 26 GBM tissue slices. (C) Heatmap depicting expression levels of high-weight genes for each metaprogram. (D) Dot plot illustrating enrichment of SSpMosaic metaprograms (columns) in 14 metaprograms (rows) defined by spatial transcriptomics in the original study. (E) Dot plot showing enrichment of SSpMosaic metaprograms (columns) in gene sets associated with programs and cell types defined by previous single-cell studies (rows). (F) Heatmap of Pearson correlation coefficients between metaprogram scores. (G) Box and scatterplots depict the TAM spot encapsulation index within MES.Hyp regions, categorizing slices into E-TAM-hyp and I-TAM-hyp groups. The nine highest encapsulation index slices (E-TAM-hyp group) are shown in (H). (I) Dot plot showing inter-region communication probabilities of ligand-receptor pairs between E-TAM-hyp and I-TAM-hyp groups. ‘‘&'' indicates co-localization between metaprograms.
Overall, by leveraging MPs to harmonize feature spaces across disparate batches, modalities, and species, SSpMosaic creates a consistent biological language within a single system and establishes a unified, interpretable, and scalable framework, eliminating the need for complex, fragmented workflows. It not only significantly lowers technical barriers and enhances standardization but also provides robust support for deciphering spatial biological mechanisms in development, homeostasis, and disease.
This study was co–first-authored by Yuelei Zhang (PhD candidate, School of Life Sciences, Nanjing University) and Wenxuan Ming (master’s student, School of Life Sciences, Nanjing University). Dr. Dijun Chen (Associate Professor, School of Life Sciences, Nanjing University) and Dr. Runzhi Deng (Associate Professor, Nanjing Stomatological Hospital Medical School of Nanjing University) served as co–corresponding authors. The source code has been openly released and is freely available for academic use and extension.
Reference: Zhang Y., Ming W., Yu B., Wang L., Lu K., Xu L., Ni Y., Deng R., Chen D. Robust integration and annotation of single-cell and spatial omics data using interpretable gene programs. Cell Genomics. 2025 Dec 19:101105. doi:10.1016/j.xgen.2025.101105.